- - English


A healthy proteome in cells is not only crucial for functionality of cells, but also essential for preventing damage (protein aggregates) that lead to a cascade of toxic events that threaten cellular health span. To ensure a proper protein homeostasis, an intricate protein quality control (PQC) network exists in cells in which Heat Shock Proteins (HSP), the central research topic in my group, play a central role.

Projectleider
Regulatie en functie Heatshock eiwitten.







PhD students:
| 2014 | Kakkar,V. (Vaishali) |
| 2014 | Minoia,M. (Melania) |
| 2013 | Rembacz,K.P. (Krzysztof) |
| 2012 | Yang,J. (Jing) |
| 2012 | Ke,L. (Lei) |
| 2011 | Zijlstra,M.P. (Marianne) |
| 2011 | Seidel,K. (Kay) |
| 2009 | Vos,M.J. (Michel) |
| 2009 | Nováková,A. (Alena) |
| 2008 | Hageman,J. (Jurre) |
| 2008 | Bosveld,F. (Floris) |
| 2007 | Yi,X. (Xia) |
| 2007 | Rujano Maldonado,M.A. (Maria) |
| 2006 | Vries,H.I. de (Hilda) |
| 2005 | Hut,H.M.J. (Hendrika) |
Disturbed protein homeostasis is a key event during cellular aging. Genetic mutations in proteins that increase the risk of protein aggregation or that impede on the protein quality control (PQC) mechanism that maintains normal protein homeostasis lead to accelerated aging or early onset of age-associated diseases.
HSP assist in protein folding as well as protein degradation (proteasomal and autophagosomal). The central aim of my group is to understand how the 100 different human HSPs are regulated to recognize their targets and steer them towards folding or degradation and how this knowledge can be exploited to combat diseases in which this protein homeostasis is disturbed.
The main focus of my research is: what regulates the input and output of protein substrates into the so-called HSP70 machines, the central hub in the cellular PQC system.
Heat shock 70 kDa proteins (HSP70s) are ubiquitous molecular chaperones that function in a myriad of biological processes. This multitude of roles is not easily reconciled with high sequence identity of Hsp70s within and across species (Figure 1A) and the universality of the activity of HSP70s in ATP-dependent client protein-binding and release cycles and wherein client protein recognition is typically rather promiscuous.
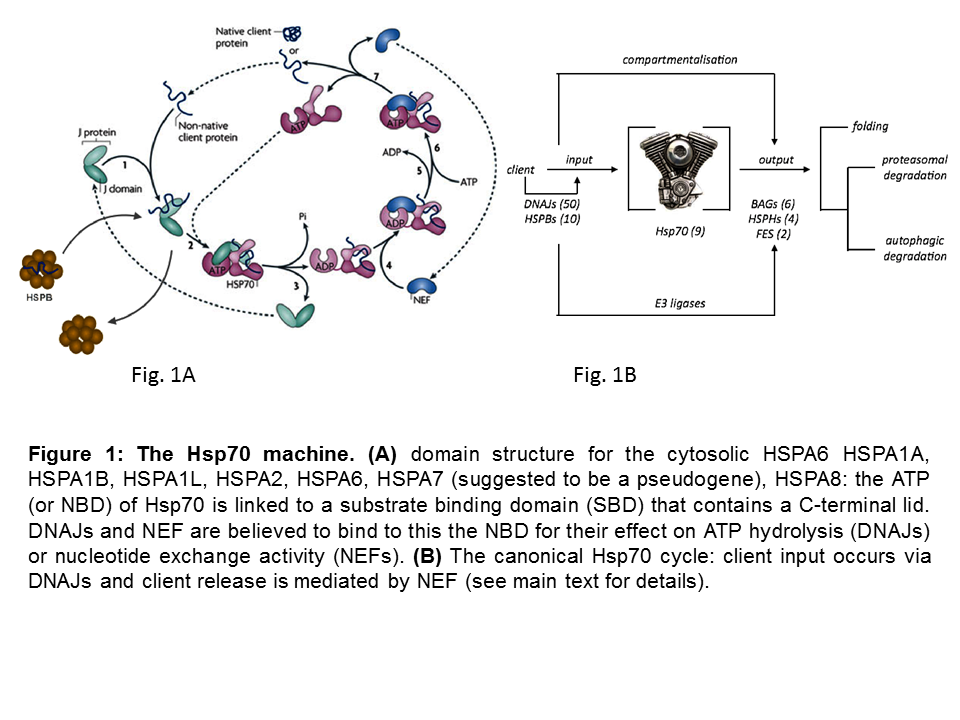
Yet, functional differences have been observed for some of the 7 cytosolic HSP70 members that are studies in my group. In addition, the apparent conundrum of multi-functionality and client binding promiscuity is likely resolved by the fact that HSP70s do not work alone, but rather as ‘HSP70 machines’, collaborating with and being regulated by several cofactors. A ‘minimal Hsp70 machine’ is made up by at least one chaperone DNAJ, one HSP70 and one Nucleotide Exchange Factor (NEF) (Figure 1B). The canonical model for the functionality of these machines has two key features: 1) the initial binding of an unfolded client protein occurs by a J protein that ‘delivers’ it to HSP70, in addition to (with the bound client) stimulating HSP70’s ATPase activity; 2) the dissociation of the client is caused by NEF actions, which gives the client either the opportunity to fold into its active conformation or, if folding is not yet complete, allow clients to re-enter the cycle such that by reiterative cycles of client binding and release productive folding can occur, whilst at the same time aggregation is prevented during the time the client is bound.
So, in this simplest view the many different DNAJs may define client specificity. At the same time the different NEFs may, besides the biochemical state of the client itself, determine the fate of the client. Figure 1C schematically summarizes our current hypothesis which is being tested in diverse projects in the lab.
]]
A myriad of age-related diseases, mostly in tissues with low regenerative capacity (brain, heart, kidney), have been associated with the existence of protein aggregates. Research from (often rare) genetic variants that predispose for such diseases have shown that the mutated proteins are not only found in the aggregates in the idiopathic cases, but also often reveal an increase in the aggregation propensity of the respective protein, strongly suggesting that protein aggregation drives the diseases pathology. This is supported by a wealth of data from experimental models.
Combined with the evidence showing that overall protein quality control declines with ageing, it has been postulated that protein homeostasis imbalances are key to the chronological ageing of many differentiated cell types (idiopathic disease) and lead to earlier onset of such diseases in case of genetic mutations.
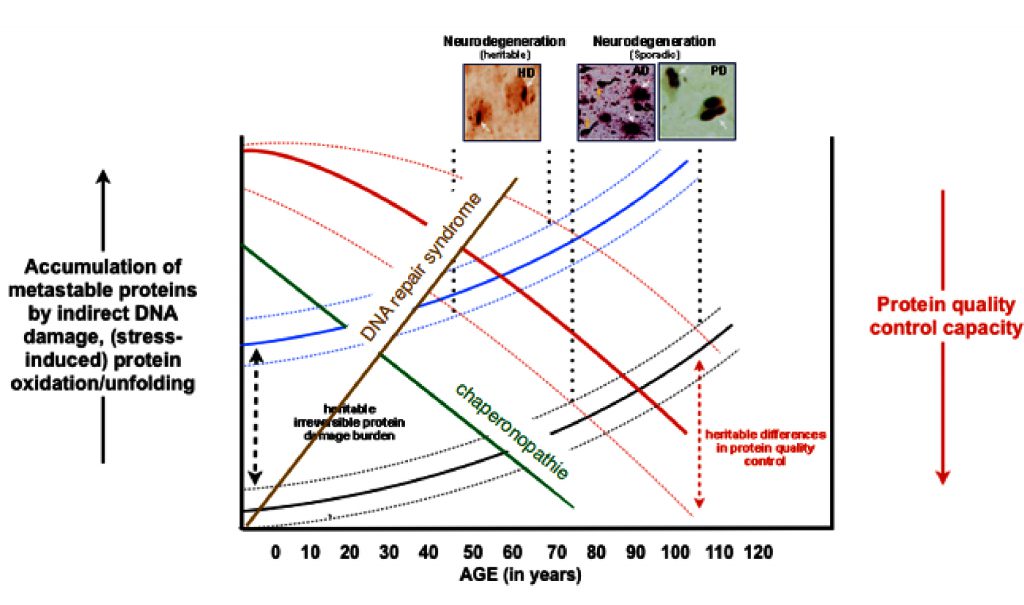 Figure 2: The hypothetical concept of protein homeostasis in aging.
Figure 2: The hypothetical concept of protein homeostasis in aging.
In my group a number of (genetic) protein aggregation diseases are studied, with the aim to identify the key, rate limiting HSPs that can counteract the respective aggregations in these diverse diseases. The diseases we have studies or are studying include:
My group has shown that, despite the fact that all these diseases are characterized by aggregates, different nodes in the HSP70 machines seem crucial to combat the respective aggregations. In particular, we have identified two structurally related DNAJ family members (DNAJB6 and DNAJB8) to be highly effective in suppressing aggregation and toxicity of the polyQ proteins causing CAG-repeat diseases (in vitro and in vivo) and of Aß-42 causing AD (in vitro).
]]
Mutations in HSPs themselves can lead to pathological conditions termed chaperonopathies that also are mostly associated with accumulation of protein aggregates. The first chaperonopathies found concerned mutations in members of the family of small HSPs (HSPBs). Later, members of the chaperonins and DNAJ families as well as some other chaperone cofactors (e.g., in BAG3, a HSP70-NEF) have been implicated heritable chaperonopathies. No genetic chaperonopathies are associated with the Hsp70/HSPA or Hsp90/HSPC family members, either because of functional redundancy within these families or because they are crucial to the central chaperone machinery such that mutations leading to functional defects are incompatible with life.
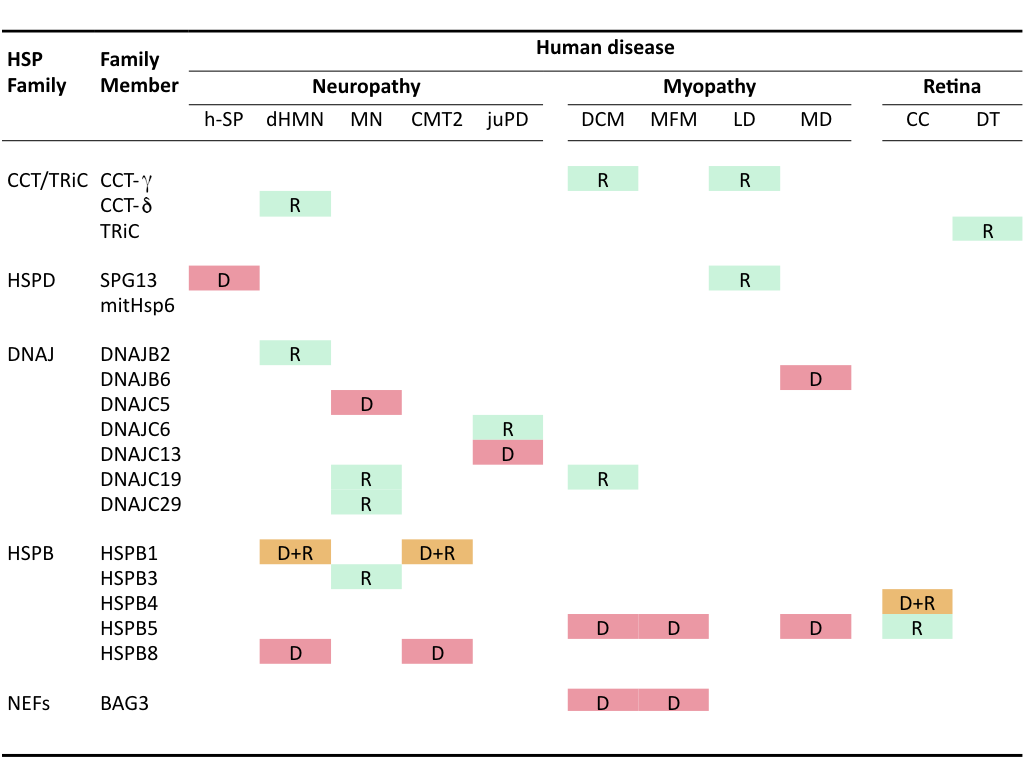
Clinically, these genetic chaperonopathies can be categorized into neuropathies (hereditary spastic paraplegia, motor neuropathy, distal hereditary motor neuropathy, dHMN), myopathies (dilated cardiomyopathy, leukodystrophy, desmin-related myopathy, mitochondrial myopathy, muscular dystrophy) or retina- and eyelens- related diseases (congenital cataracts) (Macario et al., 2005)(Table I). A variety of these mutated chaperones have been or are studied in my research group to investigate how they lead to protein aggregation (loss of function, dominant-negative effects, self-aggregation and toxic gain-of-function) and whether and how these effects can be ameliorated.
]]
We participate in the Groningen Autophagy Group.
Ongoing projects/grants:
Dissertations supervised by Harm H. Kampinga ((co-) promotor or assessor):
2019
2018
Turcan, I. (2018). Cold cases in epidermolysis bullosa: not the usual suspects[Groningen]: Rijksuniversiteit Groningen
Sen, I. (2018). New molecular mechanisms of aging regulation. [Groningen]: University of Groningen.
2017
Meister, M. (2017). On protein quality control, myofibrillar myopathies, and neurodegeneration [Groningen]: University of Groningen
van Zomeren, K. C. (2017). α-Synuclein pathology and mitochondrial dysfunction: Studies in cell models for Parkinson’s disease [Groningen]: Rijksuniversiteit Groningen
2016
Wang, C. (2016). Biomimetic drug nanocarriers to overcome biological barriers: Inspiration from pathogen invasion [Groningen]: University of Groningen
Wiersma, M. (2016). Implications of the cardiomyocyte stress response on protein homeostasis in atrial fibrillation [Groningen]: Rijksuniversiteit Groningen
Liu, W. (2016). Orphan nuclear receptor TR4 and fibroblast growth factor 1 in metabolism [Groningen]: University of Groningen
2015
Holmberg, M. (2015). Genetic factors and analysis of protein misfolding in vivo[Groningen]: University of Groningen
Kampinga, M. A. (2015). New insights in management and prognosis in acute myocardial infarction [Groningen]: University of Groningen
Munoz Llancao, P. A. (2015). Role of EPAC in axon determination [Groningen]: University of Groningen
Popken, P. (2015). Selectivity of the yeast nuclear pore complex – probing transport in vivo [Groningen]: University of Groningen
Bispo de Jesus, M. (2015). Solid lipid nanoparticles for cancer therapy: an in vitro study in prostate cancer cells [Groningen]: University of Groningen
2014
Minoia, M. (2014). Chaperones, protein homeostasis & protein aggregation diseases[S.l.]: s.n.
Kakkar, V. (2014). DNAJ proteins: more than just “co-chaperones”: Implicaties voor eiwit-aggregatie ziektes [S.l.]: [S.n.]
Regeling, A. (2014). Gene-environment interactions in Inflammatory Bowel Disease: Emphasis on smoking and autophagy ([S.l.] ed.) [S.n.]
Meijering, R. (2014). Loss of proteostasis as a substrate for atrial fibrillation: Defining novel targets for therapy [S.l.]: s.n.
Wiegman, E. M. (2014). Prediction and prevention of radiation induced lung toxicityGroningen: s.n.
2013
Jezierska, J. (2013). Genetic and molecular mechanisms underlying spinocerebellar ataxias Groningen: s.n.
2012
Yang, J. (2012). Autophagy, FOXO1 and proteostasis: implications for human diseases Groningen: s.n.
Ke, L. (2012). Molecular remodeling in atrial fibrillation: protective roles of small HSPsGroningen: s.n.
2011
Seidel, K. (2011). Neurogenerative diseases and the Protein quality control system[S.n.]
Zijlstra, M. P. (2011). The role of heat shock proteins in polyQ disorders Groningen: s.n.
2010
Rana, A. (2010). The role of impaired de novo Coenzyme A biosynthesis in pantothenate kinase-associated neurodegeneration: insight from a Drosophila modelGroningen: s.n.
2009
Vos, M. (2009). Small heat shock proteins: Implications for neurodegeneration & longevity s.n.
2008
Bosveld, F. (2008). Physiological implications of impaired de novo Coenzyme A Biosynthesis in Drosophila melanogaster s.n.
Lombaert, I. M. A. (2008). Regeneration of irradiated salivary glands by stem cell therapy Groningen: s.n.
Hageman, J. (2008). The human HSP70/HSP40 chaperone family: a study on its capacity to combat proteotoxic stress Groningen: s.n.
Burlage, F. R. (2008). The role of pilocarpine in the reduction of radiation induced damage to the salivary glands [S.n.]
2007
Rujano Maldonado, M. A. (2007). Protein quality control pathways in polyglutamine diseasesGroningen: [S.n.]
Yi, X. (2007). The impact of impaired DNA damage responses on cells, tissues and organismsGroningen: s.n.
2006
Vries, H. I. D. (2006). Grapes- and stonewall-related DNA damage responses in Drosophila melanogaster [S.n.]
Wachters, F. M. (2006). Therapeutic strategies in advanced non-small-cell lung cancer [S.n.]
2005
Hut, H. M. J. (2005). Centrosomes and cellular stress responses s.n.
1995
Stege, G. J. J. (1995). Hyperthermia and protein aggregation: role of heat shock proteins Groningen: s.n.
1989
Kampinga, H. H. (1989). Heat-induced alterations in the cell nucleus: relation to hyperthermic cell killing and radiosensitization s.n.
Information will follow.
HBO students need to have an average of 7,5




Update your browser to view this website correctly. Update my browser now

