
Deposition of protein aggregates is the hallmark of an increasing number of diseases, including neurodegenerative disorders as well as type II diabetes, peripheral amyloidoses and cardiovascular disease. How exactly the amyloid deposits are linked with cytotoxicity and neuronal death has remained enigmatic. We are using biochemical and cell biological approaches to gain insight into the molecular basis of neurodegenerative disorders associated with aberrant protein folding, such as Alzheimer’s, Parkinson’s and Huntington’s disease. Our long-term goal is to understand the mechanisms by which protein misfolding and aggregation cause cellular toxicity and how molecular chaperones and cellular stress response pathways are involved in this process.
n/a
To maintain protein homeostasis (or proteostasis) – the state of proteome balance – mammalian cells must ensure that more than 10,000 different proteins fold and assemble efficiently upon synthesis and preserve their functionally active states in a wide range of environmental and metabolic conditions. Cellular proteostasis is controlled by a complex network of factors, including molecular chaperones, proteases, and their regulators. Misfolded proteins are recognized and either refolded, degraded, or sequestered to distinct cellular sites. However, when these proteostasis machineries become compromised, as is increasingly the case during aging, aberrant proteins tend to accumulate as toxic aggregate species. This process is associated with numerous neurodegenerative diseases and other disorders, however the questions of what features make an aggregate toxic and how an aggregate harms the cell are still unresolved.
The functional proteostasis network antagonizes the build-up of aggregates or strives to neutralize their toxic effects. This may be achieved by fundamentally different strategies. Prevention of aggregation is a primary function of molecular chaperones and is achieved by binding of aggregation-prone folding intermediates, followed by their refolding or degradation. Pre-existing aggregates may be shielded by chaperones to block harmful interactions or may be removed by disaggregation or autophagy. Alternatively, toxic aggregate species, may be actively converted into large inclusion bodies, thereby reducing their reactive surfaces for toxic interactions with cellular components, or be temporarily stored in liquid like compartments like the nucleolus.
The proteostasis machinery in the nucleus differs from that of the cytosol in that no protein synthesis occurs in this compartment. In contrast to protein transport into the ER or mitochondria, the nuclear pore complexes allow the import of proteins in their folded and assembled states. Besides specific roles in histone remodeling, the nuclear chaperone machinery is therefore mainly involved in conformational protein maintenance and in the degradation of misfolded proteins. During stress, import of most proteins into the nucleus is reduced but additional chaperones and proteasome complexes enter using specific import factors. Diseases associated with protein aggregation, such as Huntington’s Disease are often characterized by the presence of intranuclear inclusions. This may be explained by recent observations that misfolded cytosolic proteins, including mutants of huntingtin, are transported into the nucleus for proteasomal degradation. When the nuclear proteostasis capacity is exhausted, misfolded proteins may then form intranuclear inclusions. Because the autophagic machinery has no access to the nucleus, perhaps the only possibility to remove these aggregates is to transport them to the cytosol after disassembly of the nuclear envelope during mitosis. This would help to explain why postmitotic cells such as neurons are more vulnerable to intra- nuclear inclusions.
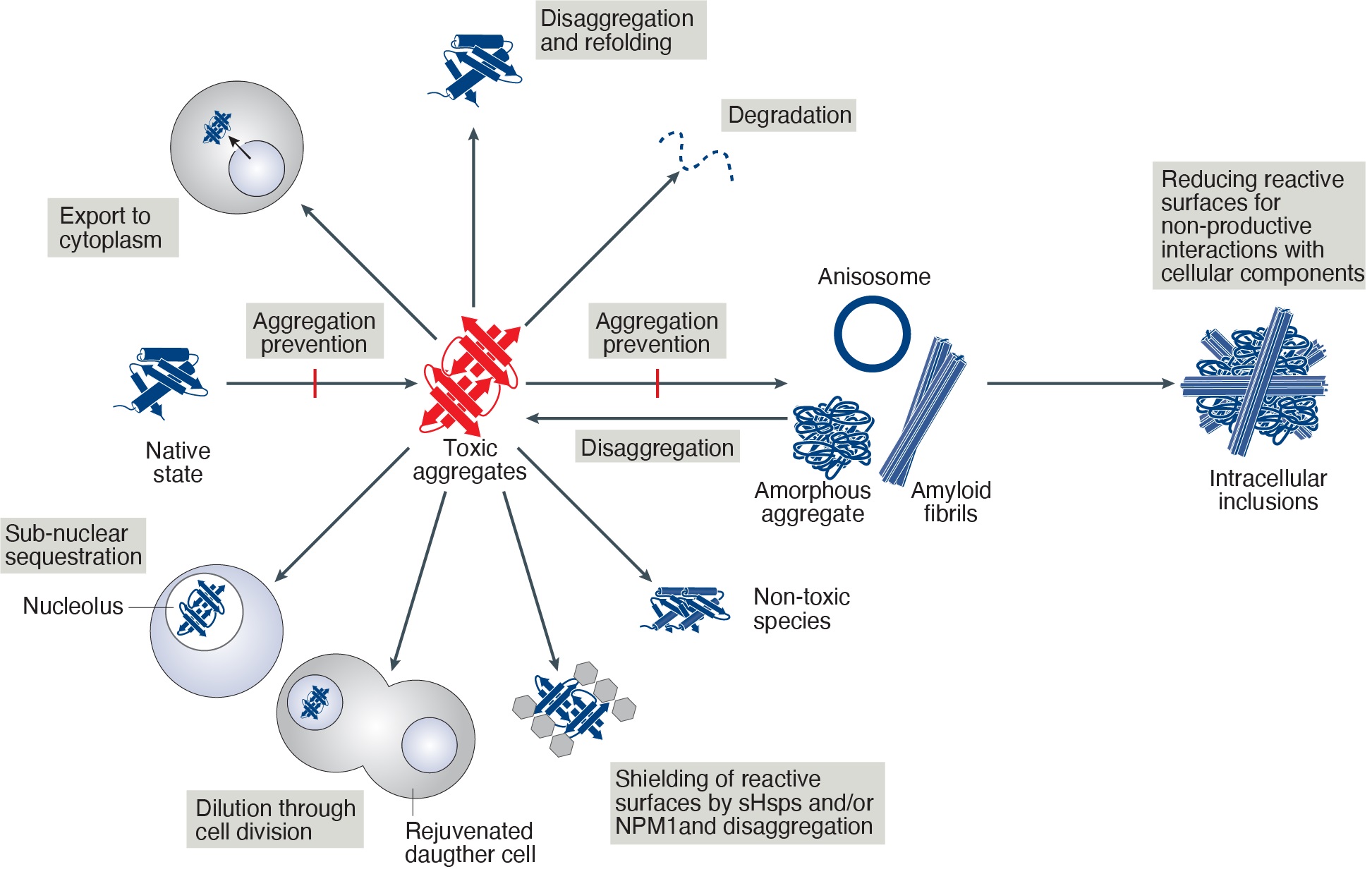
Mechanisms to counteract toxicity of nuclear aggregates. Summary of suggested pathways to handle toxic aggregates. Note that the identity of the toxic aggregate species (red) is still unknown.
Application form for an internship at one of the research groups of the section Medical Cell Biology of the department Biomedical Sciences of Cells and Systems



Update your browser to view this website correctly. Update my browser now








